


")
Our Hotline+603 - 8230 0300
Medical Device Recall
What Is Medical Device Recall?
A recall is a method of removing medical device that are in violation of laws administered by Medical Device Authority (MDA). Recall action has been set out in Section 42 of Act 737. Recall may be undertaken voluntarily and at any time by manufacturers and establishments, or at the request of the MDA.
Mandatory Recall
Mandatory recall is initiated by the authority to order for a medical device recall if a medical device possesses a high public health risk from the market as described in section 42 (4) of Act 737.
Voluntary Recall
Voluntary recall is an action taken by establishment where they are required
- to remove the medical device from the market
- to retrieve the medical device from any person to whom it has been supplied
- to notify its affected person of its defectiveness or potential defectiveness, after becoming aware that the device:
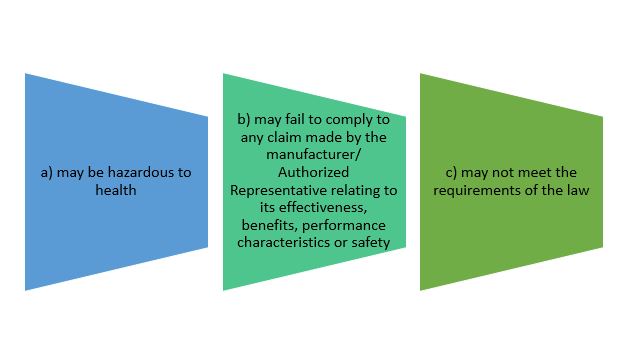
Establishment must notify authority and affected person according to recall classes before commencing recall process.
- Recall Class I, high risk, 48 hours
- Recall Class II, moderate risk, 3 days
- Recall Class III, low risk, 5 days
For further information, kindly refer to the guidance document. MDA/GD/0015 Medical Device Recall.
How To Notify Medical Device Recall?
Medical device recalls for devices that affected Malaysia can be reported via an online system, Medical Device Centralized Reporting System, MeDCReSt.
Contact Information
Vigilance Unit
MEDICAL DEVICE AUTHORITY
Ministry of Health Malaysia
Level 6, Prima 9, Prima Avenue II
Block 3547, Persiaran APEC
63000 Cyberjaya, Selangor
MALAYSIA
T: (03) 8230 0245
Updated: 20th February 2023
Field Corrective Action (FCA)
What is Field Safety Corrective Action?
Field safety corrective action (FSCA) is a post market requirement that need to be followed by an establishment under Section 41 of Act 737. The establishment is required to take proactive steps to inform the authority as well as their customers/end-users on the corrective actions made to ensure compliance of these requirements.
Manufacturers or their representatives need to carry out corrective or preventive activities in relation to their medical devices. This corrective action can be initiated through the establishment post market information such as medical device’s complaints, incident reports and so on. It also includes the field corrective actions taken by the manufacturer to reduce the risk of harm to the patient, the operator or others and/or to reduce the re-occurrence of the incident.
Field corrective action is an activity of action taken by the establishment to reduce the risk of incidents to enhance the safety and performance of a medical device. These actions may include:
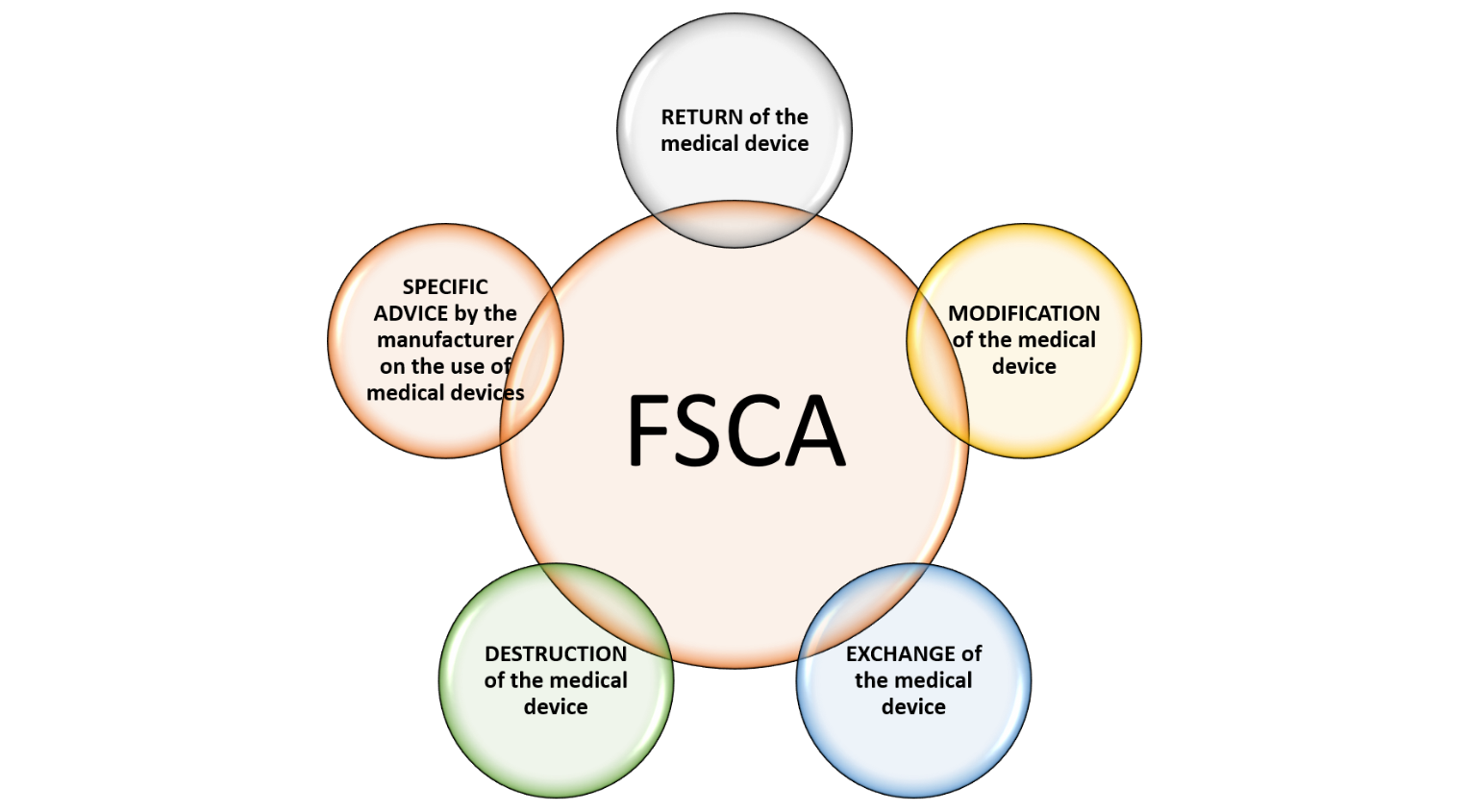 FIELD SAFETY NOTICE (FSN) is an important means of communicating a field safety corrective action (FSCA) and related safety information to users.
FIELD SAFETY NOTICE (FSN) is an important means of communicating a field safety corrective action (FSCA) and related safety information to users.
Process Flow of FSCA
For further information, kindly refer to the guidance document. MDA/GD/0013 Field Corrective Action.
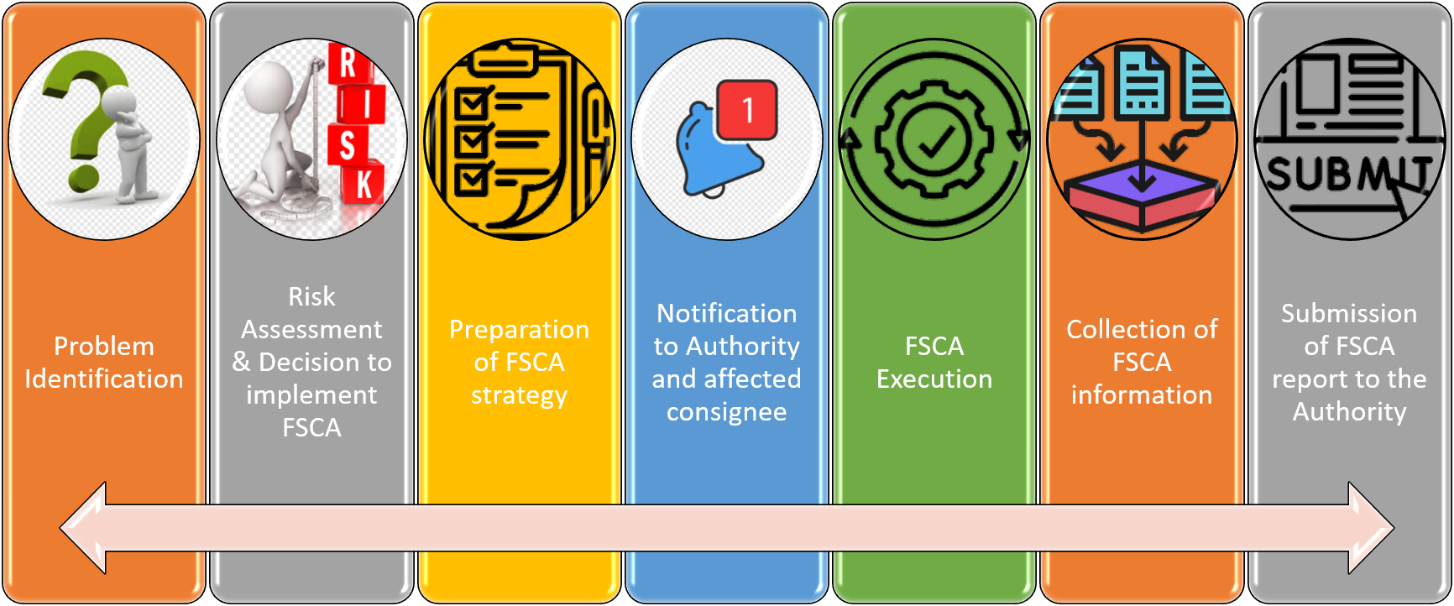
How To Notify FSCA?
Field safety corrective action (FSCA) may be requested by the Medical Device Authority for specific cases such as complaints and mandatory problem reporting (MPR). Field safety notice should be presented to the user and the Medical Device Authority before or during corrective action is taken by the establishments.
Field Safety Corrective Action (FSCA) for devices that affected Malaysia can be reported via an online system, Medical Device Centralized Reporting System, MeDCReSt
Contact Information
Vigilance Unit
MEDICAL DEVICE AUTHORITY
Ministry of Health Malaysia
Level 6, Prima 9, Prima Avenue II
Block 3547, Persiaran APEC
63000 Cyberjaya, Selangor
MALAYSIA
T: (03) 8230 0203
Updated: 20th February 2023
Mandatory Problem Reporting
What is the Mandatory Problem Reporting?
Mandatory problem reporting is also referred to as adverse event reporting in the ASEAN Medical Device Directive (AMDD).
According to Section 40 of the Act 737, the Authority shall be reported of any incident related to medical device that comes to the establishment’s attention, whether it occurs inside or outside of Malaysia.
The Medical Device Regulations 2019 define an incident as: an event that causes, or has the potential to cause, unexpected or unwanted effects involving the safety of any person who used a medical device or any person associated with the use of the medical device.
Reporting criteria
Establishments is a tendency to report when there is doubt about the reportability of an incident. Any incident that meets the three basic reporting criteria listed below is considered reportable. These criteria are;
- that an incident has occurred;
- the medical device is associated with the incident;
- the incident led to one of the following outcomes: serious deterioration in state of health, death of a patient, user, or other person, a serious threat to public health, or no death or serious injury.
There are several conditions where reporting is not required under the vigilance system for medical devices. For further information, kindly refer to the guidance document. MDA/GD/0014 Mandatory Problem Reporting.
Incident Reporting Timeline
An establishment shall report any incident that is related to the failure of a medical device or a deterioration in its effectiveness, or has led to the death or serious deterioration in the state of health of a patient, user or other person.
An initial report shall be made within thirty days of the discovery, ten days from the discovery, or forty-eight hours from the discovery.
An investigation report shall be submitted to the Authority within 30 days after the submission of an initial report. The Authority may grant an extension time to the establishment if requested.
How to report Mandatory Problem Reporting (MPR)?
MPR can be reported online via the Medical Device Centralized Reporting System, MeDCReSt
MPR for combination products must be done manually by emailing mpr@mda.gov.my and using the MPR Form: PART 1 & PART 2
Contact Information
Vigilance Unit
MEDICAL DEVICE AUTHORITY
Ministry of Health Malaysia
Level 6, Prima 9, Prima Avenue II
Block 3547, Persiaran APEC
63000 Cyberjaya, Selangor, MALAYSIA
T: (03) 8230 0213/ 0352 / 0255
Helpdesk: medcrest.mda.gov.my/pmsv_helpdesk
Updated : 20th February 2023
×



