


")
Our Hotline+603 - 8230 0300
Class A
Di bawah class A ini anak - anak nya:
-HOW TO REGISTER
-GROUPING
-CLASSIFICATION
-DIRECTIVE & CIRCULAR
IN-VITRO DIAGNOSTIC DEVICE
For any enquiries, kindly contact Registration Unit:
- Email: registration@mda.gov.my
- Phone Number: Registration Unit +603 8230 0376 or Pn. Aidahwaty bt Ariffin +603 8230 0341
- Overview of Medical Device Registration
- What is a medical device?
- Rules of Classification for General Medical Device
- Grouping for General Medical Device
- Conformity Assessments
- Declaration of Conformity
- Information on Fee Structure
According to Section 2 of Act 737, “medical device” means any instrument, apparatus, implement, machine, appliance, implant, in vitro reagent or calibrator, software, material or other similar or related article:
a. intended by the manufacturer to be used, alone or in combination, for human beings for one or more of the specific purpose (s) of;
- diagnosis, prevention, monitoring, treatment or alleviation of disease;
- diagnosis, monitoring, treatment, alleviation of or compensation for an injury;
- investigation, replacement, modification, or support of the anatomy or of a physiological process;
- supporting or sustaining life;
- control of conception;
- disinfection of medical devices;
- providing information for medical or diagnostic purposes by means of in vitro examination of specimens derived from the human body; and
which does not achieve its primary intended action in or on the human body by pharmacological, immunological or metabolic means, but which may be assisted in its intended function by such means; and
b. any instrument, apparatus, implement, machine, appliance, implant, in-vitroreagent or calibrator, software, material or other similar or related article, to be used on the human body, which the Minister may, after taking into consideration issues of public safety, public health or public risk, declare to be a medical device by order published in the
For further information, refer Guidance Document on Definition of Medical Device
Overview of the Regulatory Framework

- To address public health & safety issues
- Assessment of safety and effectiveness of medical devices
- Necessary information to allow public to make informed choices on medical devices
- Usage of certain medical devices
- Monitoring of safety and performance of medical devices in the market
- To facilitate medical device trade & industry
- Growth of medical device industry
- Local manufacturers to enter global market
Section 5(1) of Medical Device Act 2012 (Act 737) requires a medical device to be registered under the Act before it can be imported, exported or placed in the market. For that purpose, an application for the registration of a medical device must be made according to the requirement under Act 737 and in the manner determined by the Authority in Medical Device Regulation 2012.
Who is responsible to register a medical device?
- Manufacturer of a medical device as defined in Section 2 of Act 737; or
- An authorised representative appointed by a manufacturer having a principal place of business outside Malaysia, as defined in Section 2 of Act 737.
Overview of In-Vitro Diagnostic (IVD) Medical Device Registration
Class A In-Vitro Diagnostic (IVD) Medical Device
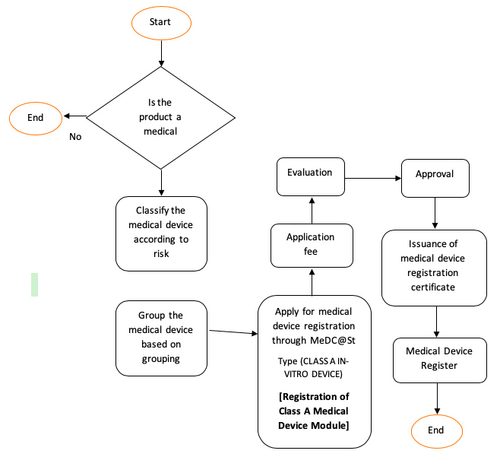
Class B, C dan D In-Vitro Device (IVD)

For further information, refer Guidelines on How to Apply for Medical Device Registration Under Medical Device Act 2012 (Act 737).
Rules of Classification for In-Vitro Diagnostic (IVD) Device

A classification of medical devices based on risk associated with the vulnerability of the human body, the technical design and the manufacture of the medical device
It uses a set of classification rules based on:
- Intended use
- The technical / scientific / medical expertise of the intended user (lay person or healthcare professional)
- The importance of the information to the diagnosis (sole determinant or one of several), taking into consideration the natural history of the disease or disorder including presenting signs and symptoms which may guide a physician;
- The impact of the result (true or false) to the individual and / or to public health.
|
Class |
Risk Level |
Device examples |
|
A |
Low |
Clinical Chemistry Analyser, Prepared Selective Culture Media |
|
B |
Low-Moderate |
Vitamin B12, Pregnancy Self-Testing, Anti-Nuclear Antibody, Urine Test Strips |
|
C |
High-Moderate |
Blood Glucose Self-Testing, HLA Typing. PSA Screening, Rubella |
|
D |
High |
HIV Blood Donor Screening, HIV Blood Diagnostic |
For further information, refer Guidance Document on Rules of Classification for General Medical Devices
Grouping for In-Vitro Diagnostic (IVD) Device
An application to register IVD devices may be made according to their grouping. For IVD devices, they may be grouped into one of the following categories as:
- Single
- Family
- System
- Set
- Test Kit
- IVD Cluster
There are 3 basic rules to determine the grouping of medical devices:
- One generic proprietary name;
- One manufacturer; and
- One common intended purpose.
For further information, refer Guidance Document on Product Grouping
Conformity Assessments is a systematic examination of evidence generated and procedures undertaken by the manufacturer under the requirements established by the Regulatory Authority to determine that a medical device is safe and performs as intended by the manufacturer and, therefore, conforms to the Essential Principles of Safety and Performance for Medical Devices.
Elements of Conformity Assessment (CA) for the purpose of medical device registration shall comprise of the following elements:
- CA of quality management system (QMS);
- CA of post-market surveillance system (PMS);
- CA of technical documentation; and
- Declaration of conformity (DoC)

Conformity assessment elements apply to Class A, B, C and D devices. According to Medical Device (Exemption) Order 2016, Class A medical devices are exempted from conformity assessment procedures by a Conformity Assessment Body under Section 7 of the Act.
Level of Conformity Assessment is proportional to the risk associated with a medical device (risk-based classification).
For further information, refer Guidance Document on Conformity Assessment, Guidance Document on Common Submission Dossier Template (CSDT) and Guidance Document on The Essential Principles of Safety and Performance of Medical Devices

Conformity Assessment Procedures for Medical Devices Approved by Recognised Countries
Section 7 of Act 737 requires a medical device to undergo conformity assessment by a registered CAB prior to its registration. However, many medical devices have undergone conformity assessment and approved in countries recognized by MDA
MDA Circular Letter No 2/2014 sets the policy relating to conformity assessment for medical devices approved by countries recognized by MDA. The policy simplifies the process of conformity assessment and accelerate medical device registration under Act 737
Policy relating to conformity assessment for medical devices approved by countries recognized by MDA
- To recognize conformity assessment and approval of medical devices placed in the market of recognized countries
- Such medical devices need to undergo a simplified conformity assessment, ie through the process of verification of evidence obtained from the manufacturer (verification process)
- The manner on how verification process shall be conducted is detailed out in Appendix 1 of the Circular Letter
For further information, refer Circular Letter of the Medical Device Authority No.2 Year 2014: Conformity Assessment Procedures for Medical Device Approved By Recognised Countries
The manufacturer shall attest that its medical device complies fully with all applicable EPSP and draws up a written Declaration of Conformity (DoC). The DoC should contain;
- Name and address of manufacturer;
- Particulars of medical device, classification;
- Name, position and signature of the responsible person who has been authorized to complete the DoC on behalf of the manufacturer; and
- CAB reviews and confirms the adequacy of the DoC by examining the supporting documents or other evidence including test report/result, applicable standards to show compliance.
Further information, refer Guidance Document on Declaration of Conformity.
As prescribe in the Fifth Schedule of the Medical Device Regulation; the application fee is imposed as follows;
Application Fee for Medical Device Registration
|
Class |
Fee Payable (RM) |
|
A |
100 |
|
B |
250 |
|
C |
500 |
|
D |
750 |
Registration Fee for Medical Device Registration
|
Class |
Fee Payable (RM) |
|
A |
- |
|
B |
1,000 |
|
C |
2,000 |
|
D |
3,000 |
|
A medical device that contains a medicinal product |
5,000 |
For further information, refer Medical Device Regulations 2012
Related Act and Regulations:
Relate Circular:
- Circular Letter of the Medical Device Authority No.2 Year 2014: Conformity Assessment Procedures for Medical Device Approved by Recognised Countries
- Medical Device (Exemption) Order 2016
Related Guidance Documents:
- How to Apply for Apply for In-Vitro Doagnostic (IVD) the Medical Device Registration under Act 737
- Definition of Medical Device
- In-Vitro Diagnostic (IVD) Medical Device Classification System
- Product Grouping
- Principles of Conformity Assessment for In- Vitro Diagnostic (IVD) Medical Devices
- Common Submission Dossier Template (CSDT) of In- Vitro Diagnostic (IVD) Medical Device
- Essential Principles of Safety and Performance of IVD Medical Devices
- Declaration of Conformity (DoC)
Updated: 20th July 2022
General Medical Device
For any enquiries, kindly contact Registration Unit:
- Email: registration@mda.gov.my
- Phone Number : Registration Unit +603 8230 0376 or Pn. Aidahwaty bt Ariffin +603 8230 0341
Decision Mesyuarat Jawatanku
- Implementation Requirements on Quality Management System (QMS) and Traceability Form
Mesyuarat Jawatankuasa Teknikal Pengkelasan dan Pendaftaran 2024 (JKTPP) Calendar (Click here)
- What is a medical device?
- Overview of the Regulatory Framework
- Overview of Medical Device Registration
- Rules of Classification for General Medical Device
- Grouping for General Medical Device
- Conformity Assessment
- Declaration of Conformity
- Information on Fee Structure
What is a medical device?
According to Section 2 of Act 737, “medical device” means any instrument, apparatus, implement, machine, appliance, implant, in vitro reagent or calibrator, software, material or other similar or related article:
a) Intended by the manufacturer to be used, alone or in combination, for human beings for one or more of the specific purpose (s) of;
• diagnosis, prevention, monitoring, treatment or alleviation of disease;
• diagnosis, monitoring, treatment, alleviation of or compensation for an injury;
• investigation, replacement, modification, or support of the anatomy or of a physiological process;
• supporting or sustaining life;
• control of conception;
• disinfection of medical devices;
• providing information for medical or diagnostic purposes by means of in vitro examination of specimens derived from the human body; and
which does not achieve its primary intended action in or on the human body by pharmacological, immunological or metabolic means, but which may be assisted in its intended function by such means; and
b) Any instrument, apparatus, implement, machine, appliance, implant, in-vitroreagent or calibrator, software, material or other similar or related article, to be used on the human body, which the Minister may, after taking into consideration issues of public safety, public health or public risk, declare to be a medical device by order published in the
For further information, refer Guidance Document on Definition of Medical Device
If you are unsure whether your product falls under the definition of a medical device as specified in Section 2 of Act 737, you may apply for Product Classification (this is link BPT work process)
Overview of the Regulatory Framework
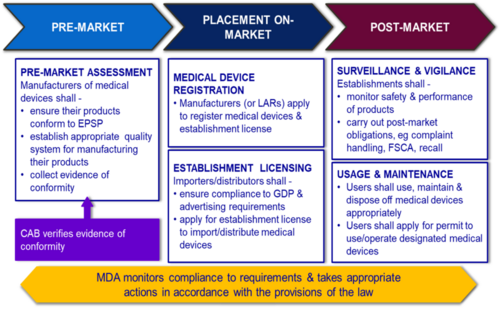
Why Regulate Medical Device?
- To address public health & safety issues
- Assessment of safety and effectiveness of medical devices
- Necessary information to allow public to make informed choices on medical devices
- Usage of certain medical devices
- Monitoring of safety and performance of medical devices in the market
- To facilitate medical device trade & industry
- Growth of medical device industry
- Local manufacturers to enter global market
Section 5(1) of Medical Device Act 2012 (Act 737) requires a medical device to be registered under the Act before it can be imported, exported or placed in the market. For that purpose, an application for the registration of a medical device must be made according to the requirement under Act 737 and in the manner determined by the Authority in Medical Device Regulation 2012.
Who is responsible to register a medical device?
- Manufacturer of a medical device as defined in Section 2 of Act 737; or
- An authorised representative appointed by a manufacturer having a principal place of business outside Malaysia, as defined in Section 2 of Act 737.
Overview of Medical Device Registration

Class A Medical Device
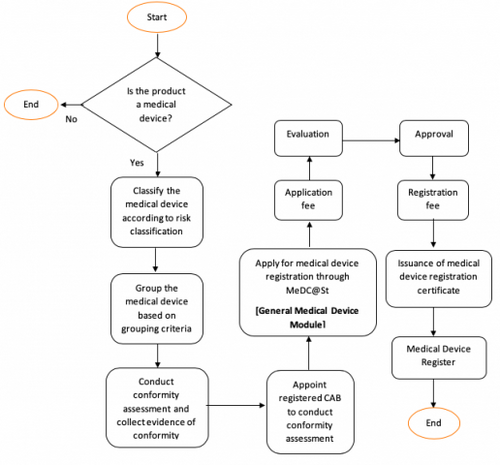
Class B, C dan D Medical Device
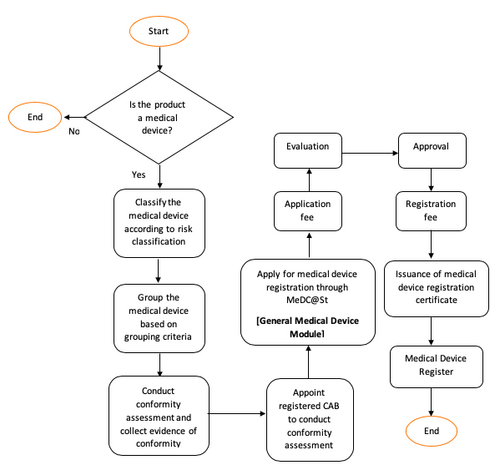
For further information, refer Guidelines on How to Apply for Medical Device Registration Under Medical Device Act 2012 (Act 737).
Rules of Classification for General Medical Device
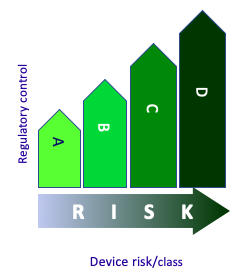
A classification of medical devices based on risk associated with the vulnerability of the human body, the technical design and the manufacture of the medical device
It uses a set of classification rules based on:
- intended use
- duration of use (transient, short-term and long-term)
- part of human body (non-invasive or invasive with respect to body orifices, surgically invasive interventions, central circulatory system, central nervous system)
|
Class |
Risk Level |
Device examples |
|
A |
Low |
Simple surgical instruments, tongue depressor, liquid-in-glass thermometer, examination light, simple wound dressing, oxygen mask, stethoscopes, walking aids |
|
B |
Low-Moderate |
Hypodermic needles, suction equipment, anesthetic breathing circuits, aspirator, external bone growth simulators, hearing aids, hydrogel dressings, patient-controlled pain relief, phototherapy unit, x-ray films |
|
C |
High-Moderate |
Lung ventilator, orthopedic implants, baby incubator, blood oxygenator, blood bag, contact lens disinfecting/cleaning products, deep wound dressing, defibrillator, radiological therapy equipment, ventilator |
|
D |
High |
Pacemakers and their leads, implantable defibrillators, implantable infusion pumps, heart valves, inter-uterine contraceptive devices, neurological catheters, vascular prostheses, stents |
For further information, refer Guidance Document on Rules of Classification for General Medical Devices
Grouping for General Medical Device
An application to register medical devices may be made according to their grouping. For general medical devices, they may be grouped into one of the following categories as:
- Single
- Family
- System
- Set
There are 3 basic rules to determine the grouping of medical devices:
- One generic proprietary name;
- One manufacturer; and
- One common intended purpose.
For further information, refer Guidance Document on Product Grouping
Conformity Assessment
Conformity Assessment is a systematic examination of evidence generated and procedures undertaken by the manufacturer under the requirements established by the Regulatory Authority to determine that a medical device is safe and performs as intended by the manufacturer and, therefore, conforms to the Essential Principles of Safety and Performance for Medical Devices.
Elements of Conformity Assessment (CA) for the purpose of medical device registration shall comprise of the following elements:
- CA of quality management system (QMS);
- CA of post-market surveillance system (PMS);
- CA of technical documentation; and
- Declaration of conformity (DoC)

Conformity assessment elements apply to Class A, B, C and D devices. According to Medical Device (Exemption) Order 2016, Class A medical devices are exempted from conformity assessment procedures by a Conformity Assessment Body under Section 7 of the Act.
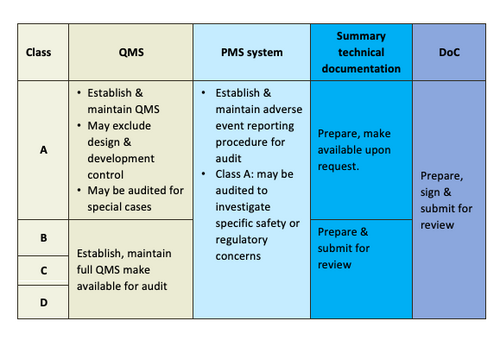
Level of Conformity Assessment is proportional to the risk associated with a medical device (risk-based classification).
For further information, refer Guidance Document on Conformity Assessment, Guidance Document on Common Submission Dossier Template (CSDT) and Guidance Document on The Essential Principles of Safety and Performance of Medical Devices
Conformity Assessment Procedures for Medical Devices Approved by Recognised Countries
Section 7 of Act 737 requires a medical device to undergo conformity assessment by a registered CAB prior to its registration. However, many medical devices have undergone conformity assessment and approved in countries recognized by MDA
MDA Circular Letter No 2/2014 sets the policy relating to conformity assessment for medical devices approved by countries recognized by MDA. The policy simplifies the process of conformity assessment and accelerate medical device registration under Act 737
Policy relating to conformity assessment for medical devices approved by countries recognized by MDA
- To recognize conformity assessment and approval of medical devices placed in the market of recognized countries
- Such medical devices need to undergo a simplified conformity assessment, ie through the process of verification of evidence obtained from the manufacturer (verification process)
- The manner on how verification process shall be conducted is detailed out in Appendix 1 of the Circular Letter
For further information, refer Circular Letter of the Medical Device Authority No.2 Year 2014: Conformity Assessment Procedures for Medical Device Approved By Recognised Countries
Declaration of Conformity
The manufacturer shall attest that its medical device complies fully with all applicable EPSP and draws up a written Declaration of Conformity (DoC). The DoC should contain;
- Name and address of manufacturer;
- Particulars of medical device, classification;
- Name, position and signature of the responsible person who has been authorized to complete the DoC on behalf of the manufacturer; and
- CAB reviews and confirms the adequacy of the DoC by examining the supporting documents or other evidence including test report/result, applicable standards to show compliance.
Further information, refer Guidance Document on Declaration of Conformity.
Information on Fee Structure
As prescribe in the Fifth Schedule of the Medical Device Regulation; the application fee is imposed as follows;
Application Fee for Medical Device Registration
|
Class |
Fee Payable (RM) |
|
A |
100 |
|
B |
250 |
|
C |
500 |
|
D |
750 |
Registration Fee for Medical Device Registration
|
Class |
Fee Payable (RM) |
|
A |
- |
|
B |
1,000 |
|
C |
2,000 |
|
D |
3,000 |
|
A medical device that contains a medicinal product |
5,000 |
For further information, refer Medical Device Regulations 2012
Related Act and Regulations:
Related Circulars:
- Circular Letter of the Medical Device Authority No.2 Year 2014: Conformity Assessment Procedures for Medical Device Approved by Recognised Countries
- Medical Device (Exemption) Order 2016
Related Guidance Documents:
- How to Apply for Medical Device Registration Under Medical Device Act 2012 (Act 737).
- Definition of Medical Device
- Rules of Classification for General Medical Devices
- Product Grouping
- Guidance Document on Conformity Assessment
- Common Submission Dossier Template (CSDT)
- The Essential Principles of Safety and Performance of Medical Devices
- Declaration of Conformity (DoC)
Updated: 7 / 8 / 2023
×



